
Xavier RAFFOUX
last updated 12/06/2023
xavier.raffoux@inrae.fr
Biologie de l'Adaptation et Systèmes en Évolution
Atelier Cartographie, Expression et Polymorphisme
INRAE, Engineer
publications - orcid
Thematics:
- Studying the genetic determinism of the number and position of meiotic recombination events and of the interactions between recombination and evolution during adaptation.
- Analysis of “Omics” data for the unit: Bioinformatics and statistics
- Conducting molecular biology projects for the unit
Training:
- PhD in Life and Health Sciences, Paris-Sud University (Orsay, France), 2018
- Master in Biology - Genome, Cells, Development, Evolution, Paris-Sud University (Orsay, France), 2013
- Diploma of Higher Education in Biology, UPMC (Paris, France), 1999-2000
Experiences
- Research technician, GQE-Le Moulon (50% ACEP, 50% RAMDAM)
- PhD student, GQE-Le Moulon (Gif-sur-Yvette, France), 2013-2018
- Research technician, GQE-Le Moulon (Gif-sur-Yvette, France), 2003-2012
- Animal technician, LBMC Laboratory of Molecular and Cellular Biology, (Lyon, France), 2002-2003
Meiosis research project
One of the team’s themes aims to study the phenomenon of meiotic recombination since it is one of the major players in the evolution of genomes. Indeed, for each gamete to contain a complete set of chromosomes, physical links must be created, during meiosis, between the two copies (homologs) of each chromosome. These links lead to a reciprocal exchange of genetic material between homologous chromosomes called crossing-over. This is how part of the genetic information, originating from two parent individuals, can be combined on the same chromosome, and therefore in a descendant who then presents a new combination of characters that did not exist in the parents. Sexual reproduction thus allows the creation of new assemblages of characters in the offspring, and this new diversity is then subject to selection, whether natural or directed by humans, for example from an agronomic point of view. In addition, many genetic studies exploit recombination events to identify regions of the genome responsible for variation in traits of interest, and the accuracy of these studies depends in part on the level of recombination. For these reasons, understanding the genetic factors controlling the number and position of recombination events is a crucial issue for medicine, agriculture, or basic research. Part of the team to which I belong therefore studies, using theoretical and experimental approaches, the formation of meiotic crossing-overs, the regulation of their number and their distribution, the relationships between recombination and evolution and finally their implications for the improvement Plant.
More specifically, the ANR EvolRec project, which began in 2021, in connection with this theme, raises the following scientific questions in the yeast S. cerevisiae:
- How does the recombination rate respond to directional selection?
- What are the genetic determinants of the number and distribution of crossing-overs?
- What are the interactions between recombination and evolution during adaptation?
As part of this project, I am continuing the work initiated during my thesis on the control of the number and position of crossing-overs and I am developing new approaches aimed at studying the relationships between recombination and adaptation via evolution. genomes. Thus, for several years I have been conducting a recurrent selection experiment on the recombination rate in S. cerevisiae in order (1) to study the response to selection for this trait, and (2) to detect recombination QTLs (Quantitative Trait Locus = region of the genome associated with the variation of a quantitative trait).
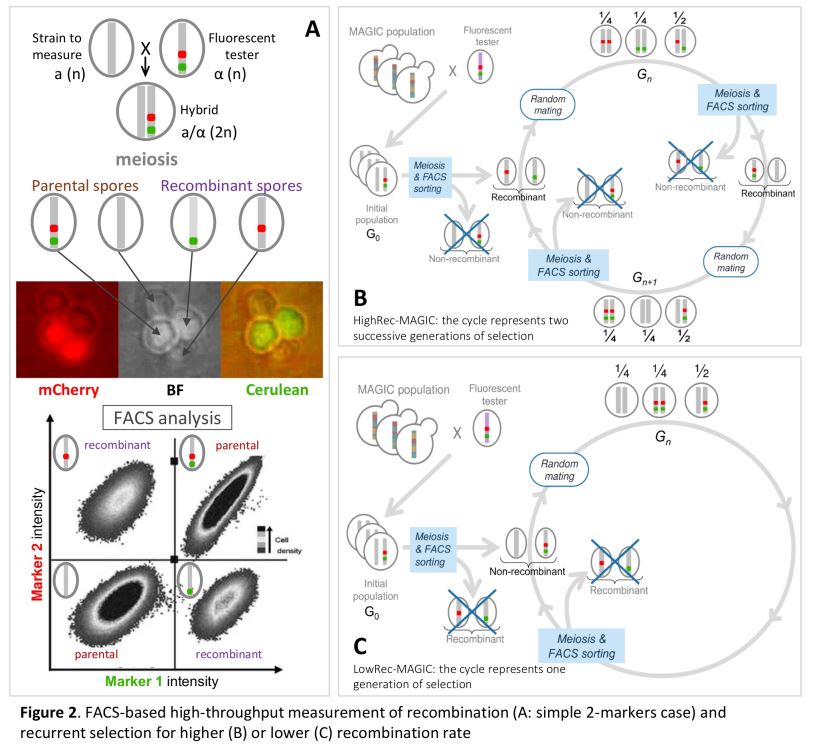
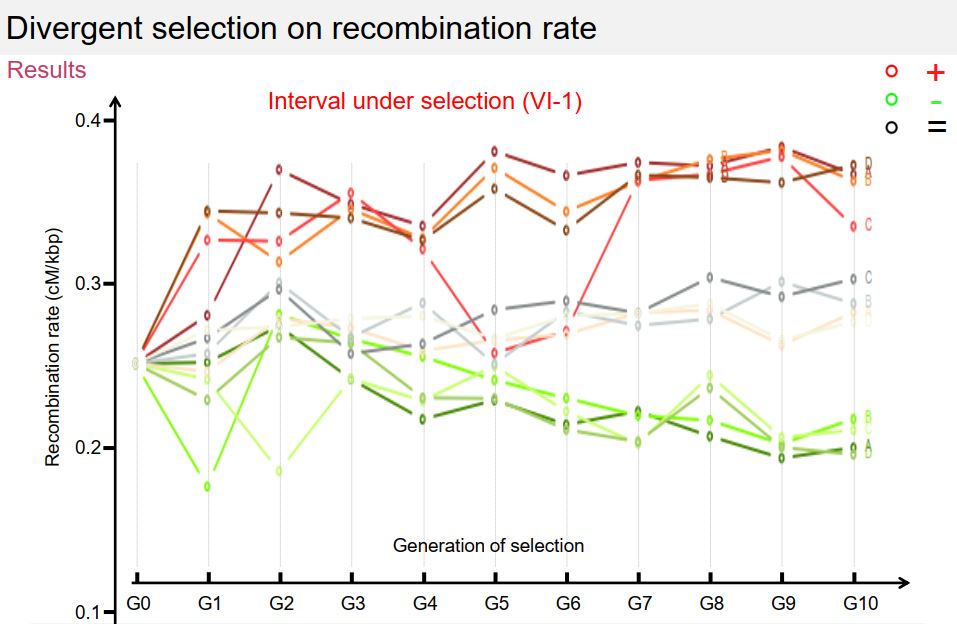
In this context, I developed an original method based on flow cytometry, allowing the high-throughput selection of spores according to their recombinant status or not between two fluorescent markers (300,000 individuals sorted in 15 minutes), then inter-cross them in panmixia to form the next generation. Thus, it is now possible to recurrently select spores according to their recombination status, which removes a technical obstacle and opens a new way for the study of the genetic determinism of the recombination rate. Thanks to this approach, I carried out 11 generations of recurrent selection according to three methods: (A) selection of individuals having recombined between the markers, (B) selection of the descendants having not recombined between the markers and (C) selection of the descendants regardless of the level of recombination between the markers. The results obtained for the 12 populations and over the 11 generations of selection show that the level of recombination varies in response to selection in the chromosomal region where the selection is applied, and that it varies in the opposite direction, and to a lesser extent. measurement, in the adjacent region, suggesting local compensation (interference) effects. On the other hand, the level of recombination does not show any long-distance variation of the interval under selection, nor on the other chromosomes studied. Finally, this variation did not appear gradually over generations, but in steps, and recombination returned to its initial level within one generation when the selection pressure was lifted. These 13 populations were sequenced at high coverage (50X) which will make it possible to detect differences in allele frequencies between the initial population and the evolved populations on the one hand, and between the 3 modes of selection on the other hand.
Still in a research theme centered on meiotic recombination, I am participating in the reflection on a work package for a second ANR project (CO-PATT), also funded from 2021, led by Éric Espagne (I2BC, Orsay).
As part of this project, we will carry out a suppressor screen in yeast to detect interference-restoring genes in mutants of the slx5 gene which reduces interference between crossing-overs.
Molecular biology projects
Estimation of the frequencies of different varieties of wheat, after sowing then harvesting in mixture
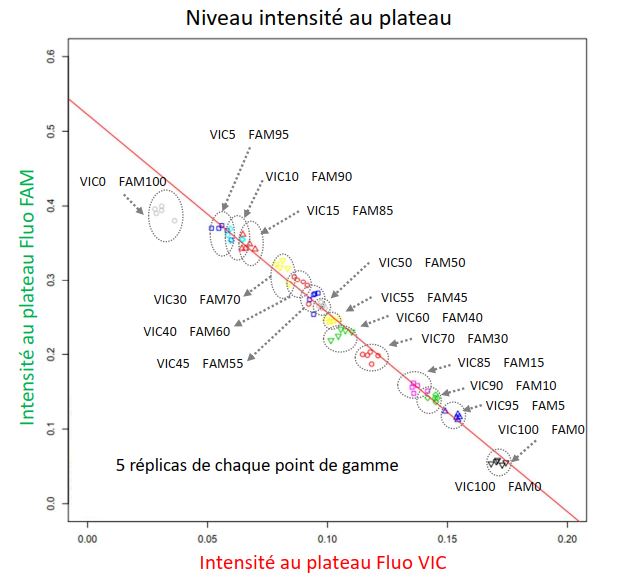
First, as part of the theme of participatory design of varietal mixtures of the Diversity, Evolution and Adaptation of Populations (DEAP) team, my objective is to estimate the frequency of different wheat varieties, after sowing then harvesting in mixture. We have chosen to design a method combining two existing molecular biology techniques: real-time quantitative PCR and competitive allele-specific PCR (KASP). I developed an R script allowing to model the data produced in order to extract certain parameters such as the efficiency of the reaction, the level of fluorescence at the plateau or the Ct, this from the raw data of fluorescence during the cycles of amplification. My work thus makes it possible to exploit the data and to estimate the frequencies of each variety from two of the proxies extracted from the amplification curve in real time (level of fluorescence at the plateau and Ct). The results, carried out on a range of binary mixtures of wheat leaf DNA in varying proportions, show that we are able to estimate these frequencies with 5% uncertainty. The objectives set are to experimentally improve the precision of the estimate and then to validate a panel of molecular markers that can be used at higher throughput.
In the short term, this work will make it possible to know the proportions of wheat genotypes present in mixtures in the plot from DNA samples from flour mixtures.
It will then be possible to study the evolution of the proportions of different genotypes in a mixture over the generations.
This is the case of experimental devices where the harvest is used to sow the following year.
Finally, in the case of agricultural practices where a new mixture of grains is prepared each year, this work will make it possible to study which factors (inputs, parasites, abiotic stress) can influence the competing crop of several genotypes.
Genotype verification before de novo sequencing
Secondly, as part of a project aiming to study how structural variations of the genome (transposable elements, gene duplications) can modulate the transcriptome, I genotyped maize lines by microsatellites to verify their identity before their sequencing of new. For this project, I led by adapting the existing technical protocols, ensured the reliability of the results (double reading system), formatted and then transmitted the results to the researchers involved during an end-of-project meeting.
Development of microsatellite markers in the hoary apple aphid
Finally, thirdly, within the framework of a project aimed at studying the adaptive coevolution between a parasite (ash aphid) and a host subject to domestication (apple tree), my objective was to be able to identify different genotypes of aphids and to verify their mode of reproduction in a culture chamber. To carry out this project, in consultation with the head of the ACEP team, Mr. Falque, we made the choice of the technique, the markers to be used and their number (dimensioning of the experiment according to the questions asked ), this during a meeting at the start of the project with the doctoral student involved. I then produced the first data (genotyping of ash aphids on 8 microsatellites), analyzed and formatted the results.
Bioinformatics and statistical analysis of omics data
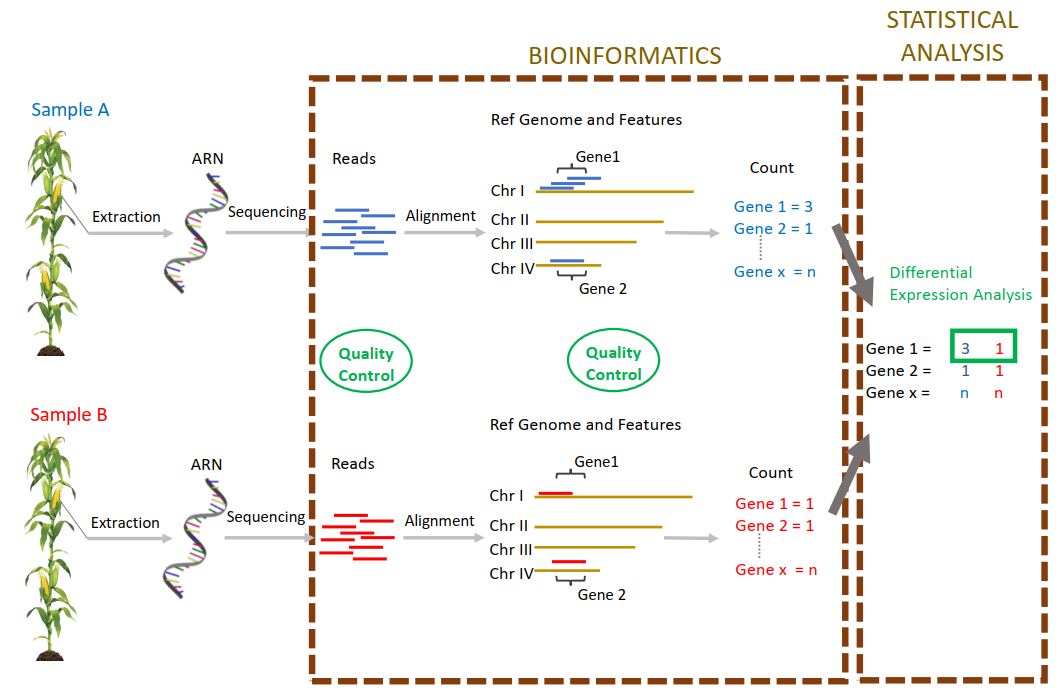
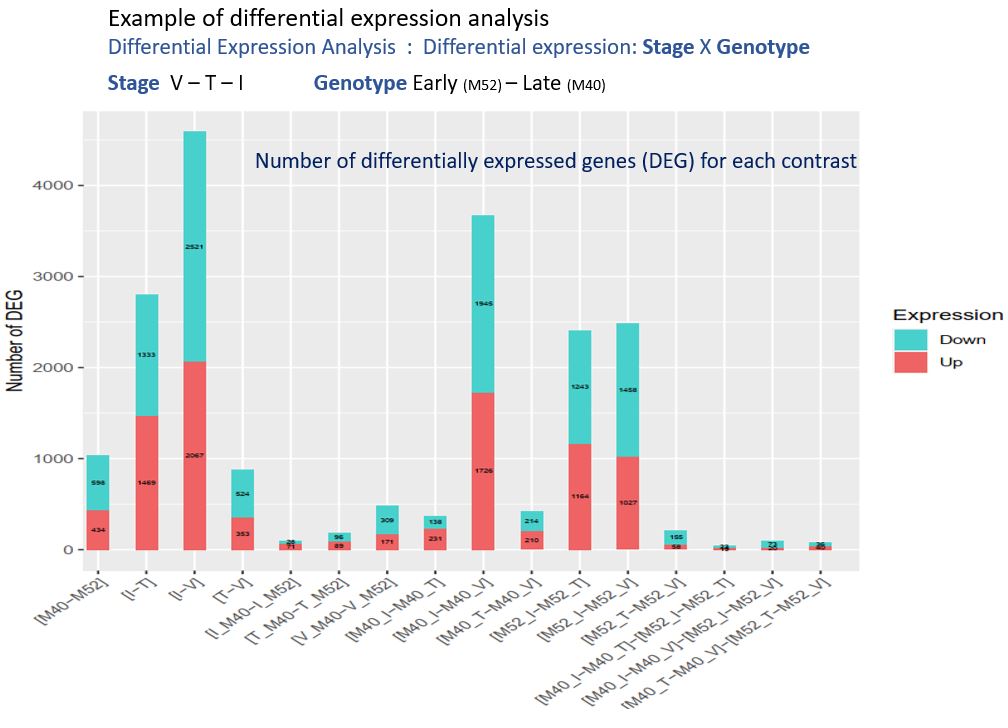
Currently, as part of one of the tasks of the “Itemaize” project which aims to study the target traits of selection for flowering date in maize, I am analysing RNAseq data from two maize genotypes. These two genotypes are the result of a divergent selection experiment on the precocity of flowering, an experiment carried out for more than 20 years in the unit. Each year, plants are selected on the basis of their flowering date, to form the next generation. My objective is to study the level of gene expression in the meristems of plants of these two genotypes (early – late), collected at generation 18 and at different stages of development. Thus, it is possible to detect genes differentially expressed between early and late genotypes, between developmental stages, and to study the interactions between these two factors. The genes detected are potentially involved in floral transition, a character of major agronomic interest in the context of climate change. Further downstream, the study of the response to selection during this experiment and of the different evolutionary paths followed will inform us about the evolutionary processes involved in the context of adaptation to a new environment.
After a quality control of the data (FastQC and MultiQC), in order to detect possible sequencing problems or experimental biases, I aligned the reads on the reference genome. For this, I chose an adapted alignment software (STAR), designed to perform this task on complex genomes such as maize. This is indeed a eukaryotic genome with a size of 5,000 Mbp, with the presence of duplicated genes and transposable elements. In addition, this software uses the genome annotation information to perform the alignment, but can also discover new introns not yet annotated. These data allowed me to produce a counting table (number of reads per gene and per sample) using the “HtSeqCount” software and the parameters allowing a counting which is the most accurate reflection of the level of expression of the genes. Principal component analysis on the raw count data allowed me to observe that the data is structured according to our two factors (Genotype and flowering stage) but not between the biological replicas, which makes sense in this experiment. One of the samples was excluded from the following analyses because it was too different from the others.
I then wrote an R script using the DiCoExpress library to statistically analyze the counting data and thus detect genes showing differential expression in the context of floral transition.
For this, I normalized the raw counting data (TMM method), defined a model, and a detection threshold. For each of the contrasts studied, I observed the distribution of p-values and thus verified that the threshold for the minimum number of reads per gene was relevant. I then searched the bibliography for information on the function of some of the genes detected and found information related to floral transition.
As this work could not be carried out for all the genes detected, I did an ontology work by replacing each gene in its function group.
This work makes it possible to have an overview and to understand which metabolic pathways are regulated differently at the time of the floral transition.
I will soon choose and then use statistical analyzes to continue my work on a multi-omics scale, with for example the integration of phenotyping data.
The results obtained will then be coupled with metabolomic and proteomic data in order to build a floral transition regulatory network.
Publications
2021
- Olvera-Vazquez SG., Alhmedi A., Miñarro M., Shykoff JA., Marchadier E., Rousselet A., Remoué C., Gardet R., Degrave A., Robert P., Chen X., Porchier J., Giraud T., Vander-Mijnsbrugee K., Raffoux X. , Falque M., Alins G., Didelot F., Beliën T., Dapena E., Lemarquand A., Cornille A.. (2021) Experimental test for local adaptation of the rosy apple aphid (Dysaphis plantaginea) to its host (Malus domestica) and to its climate in Europe. PCI Ecology, (Pre-registration version)
2018
- Raffoux X. , 2018-06-11 06/11/18, Diversité et déterminisme génétique de la recombinaison méiotique chez Saccharomyces cerevisiae, PhD thesis, Université Paris-Saclay
- Raffoux X. , Bourge M., Dumas F., Martin OC., Falque M.. (2018) High-throughput measurement of recombination rates and genetic interference in Saccharomyces cerevisiae. Yeast, 6 (35) 431-442
- Raffoux X. , Bourge M., Dumas F., Martin OC., Falque M.. (2018) Role of Cis, Trans, and Inbreeding Effects on Meiotic Recombination in Saccharomyces cerevisiae. Genetics, 4 (210) 1213-1226